蛋白質深度學習







在撰寫本文時,我考慮了兩個受眾。一個是正在嘗試進入機器學習領域的生物學家,另一個是正在嘗試進入生物學領域的機器學習者。如果您對生物學或機器學習都不熟悉,仍然歡迎您閱讀,但有時可能會覺得有些困惑!如果您已經對兩者都很熟悉,那麼您可能根本不需要這篇文章——您可以直接跳到我們的示例筆記本,檢視這些模型的實際應用
- 蛋白質語言模型微調(PyTorch,TensorFlow)
- 使用ESMFold進行蛋白質摺疊(目前僅限於PyTorch,因為存在
openfold依賴項)
生物學家入門:語言模型到底是什麼?
用於處理蛋白質的模型深受BERT和GPT等大型語言模型的啟發。因此,為了理解這些模型的工作原理,我們將回到2016年左右,也就是它們出現之前。唐納德·特朗普尚未當選,英國脫歐尚未發生,深度學習(DL)是每天都在創造新紀錄的熱門新技術。DL成功的關鍵在於它使用人工神經網路來學習資料中複雜的模式。然而,DL有一個關鍵問題——它需要大量資料才能良好工作,而在許多工中,這些資料根本不可用。
假設您想訓練一個DL模型,以英語句子作為輸入,並判斷其語法是否正確。因此,您收集了訓練資料,它看起來像這樣:
| 文字 | 標籤 |
|---|---|
| The judge told the jurors to think carefully. | 正確 |
| The judge told that the jurors to think carefully. | 不正確 |
| ... | ... |
理論上,這項任務在當時是完全可行的——如果您將這樣的訓練資料輸入到DL模型中,它就可以學習預測新句子是否語法正確。實際上,它的效果並不好,因為在2016年,大多數人會為他們想要訓練的每個任務隨機初始化一個新模型。這意味著模型必須僅從訓練資料中的示例中學習所需的一切!
為了理解這有多困難,假設您是一個機器學習模型,我正在為您提供一些我希望您學習的任務的訓練資料。如下:
| 文字 | 標籤 |
|---|---|
| Is í an stiúrthóir is fearr ar domhan! | 1 |
| Is fuath liom an scannán seo. | 0 |
| Scannán den scoth ab ea é. | 1 |
| D’fhág mé an phictiúrlann tar éis fiche nóiméad! | 0 |
我在這裡選擇了一種我希望你從未見過的語言,所以我想你可能對自己已經學會這項任務的信心不是很足。也許在數百或數千個例子之後,你可能會開始注意到輸入中一些重複的詞語或模式,並且你可能會做出比隨機猜測更好的猜測,但即使如此,一個新詞或不尋常的措辭肯定會讓你感到困惑,並導致你做出錯誤的猜測。這並非巧合,當時DL模型的表現也差不多就是這樣!
現在嘗試相同的任務,但用英語進行
| 文字 | 標籤 |
|---|---|
| She’s the best director in the world! | 1 |
| I hate this movie. | 0 |
| It was an absolutely excellent film. | 1 |
| I left the cinema after twenty minutes! | 0 |
現在很簡單——任務只是預測影評是積極(1)還是消極(0)。僅憑兩個積極例子和兩個消極例子,你可能就能以接近100%的準確率完成這項任務,因為**你已經擁有大量的英語詞彙和語法,以及圍繞電影和情感表達的文化背景知識。**如果沒有這些知識,情況就會更像第一個任務——你需要閱讀大量的例子才能開始發現輸入中哪怕是膚淺的模式,即使你花時間研究了成千上萬個例子,你的猜測準確率仍然遠低於在英語任務中只用四個例子時的準確率。
關鍵突破:遷移學習
在機器學習中,我們將這種將先驗知識轉移到新任務的概念稱為“**遷移學習**”。讓這種遷移學習在深度學習中發揮作用是2016年左右該領域的一個主要目標。雖然預訓練詞向量(非常有趣,但超出本文範圍!)在2016年確實存在,並允許將一些知識轉移到新模型中,但這種知識轉移仍然相對膚淺,模型仍然需要大量訓練資料才能良好工作。
這種情況一直持續到2018年,當時兩篇重要的論文相繼問世,引入了模型ULMFiT,隨後是BERT。這些是首次讓自然語言遷移學習真正發揮作用的論文,特別是BERT標誌著預訓練大型語言模型時代的開始。這兩篇論文共同的訣竅是,它們利用了深度學習中人工神經網路的內部結構——它們在一個訓練資料非常豐富的文字任務上長時間訓練了一個神經網路,然後將整個神經網路複製到新任務中,只改變了對應於網路輸出的幾個神經元。
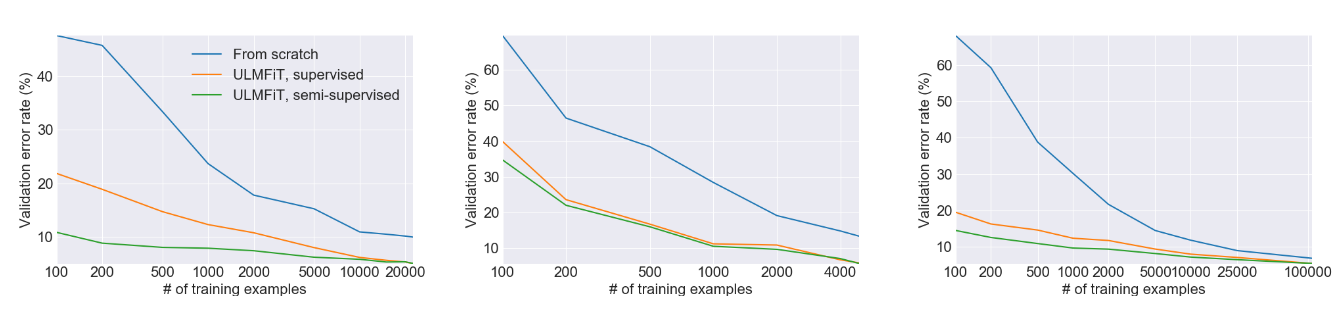
此圖來自ULMFiT論文,顯示了使用遷移學習與從頭開始訓練模型在三個獨立任務上的巨大效能提升。在許多情況下,使用遷移學習的效能相當於擁有100倍以上的訓練資料。別忘了這篇論文是在2018年發表的——現代大型語言模型甚至可以做得更好!
之所以有效,是因為在解決任何非平凡任務的過程中,神經網路學習了大量輸入資料的結構——視覺網路在給定原始畫素的情況下,學會識別線條、曲線和邊緣;文字網路在給定原始文字的情況下,學會識別語法結構的細節。然而,這些資訊並非任務特定的——遷移學習之所以有效,關鍵原因是**解決任務所需的大部分知識並非任務特有的!**要對電影評論進行分類,你不需要了解很多關於電影評論的知識,但你需要大量的英語知識和文化背景。透過選擇一個訓練資料豐富的任務,我們可以讓神經網路學習那種“領域知識”,然後將其應用於我們關心的、訓練資料可能更難獲得的全新任務。
至此,希望您已經理解了什麼是遷移學習,以及大型語言模型只是一個經過大量文字資料訓練的巨大神經網路,這使其成為轉移到新任務的理想選擇。我們將在下面看到這些相同的技術如何應用於蛋白質,但首先我需要為我的另一半受眾寫一個介紹。如果您已經熟悉,請隨意跳過這一部分!
機器學習者入門:蛋白質到底是什麼?
一句話概括:蛋白質的功能非常多。有些蛋白質是酶——它們充當化學反應的催化劑。當您的身體將營養物質轉化為能量時,從食物到肌肉運動的每一步都由酶催化。有些蛋白質是結構蛋白——它們提供穩定性和形狀,例如在結締組織中。如果您曾看過化妝品廣告,您可能見過膠原蛋白、彈性蛋白和角蛋白等詞語——這些蛋白質構成了我們皮膚和頭髮的大部分結構。
其他蛋白質在健康和疾病中至關重要——每個人可能都記得關於COVID-19病毒的**刺突蛋白**的無數新聞報道。COVID刺突蛋白與人體細胞表面的一種名為ACE2的蛋白質結合,這使其能夠進入細胞並傳遞其病毒RNA有效載荷。由於這種相互作用對感染至關重要,因此在疫情期間,對這些蛋白質及其相互作用進行建模是一個巨大的焦點。
蛋白質由多個氨基酸組成。氨基酸是相對簡單的分子,都具有相同的分子主鏈,並且該主鏈的化學性質允許氨基酸融合在一起,從而使單個分子可以形成一個長鏈。這裡關鍵要理解的是,氨基酸只有少數幾種不同型別——20種標準氨基酸,另外根據特定生物體可能還有一些稀有和奇怪的。蛋白質種類繁多的原因在於這些**氨基酸可以以任何順序組合**,並且由此產生的蛋白質鏈可以具有截然不同的形狀和功能,因為鏈的不同部分會相互粘附和摺疊。這裡可以把文字作為類比——英語只有26個字母,但想想你可以用這26個字母的組合寫出多少種不同型別的東西!
事實上,由於氨基酸種類很少,生物學家可以為每種氨基酸分配一個唯一的字母。這意味著你可以將蛋白質寫成一個文字字串!例如,假設一個蛋白質鏈中含有蛋氨酸、丙氨酸和組氨酸。這些氨基酸的對應字母就是M、A和H,因此我們可以將該鏈寫為“MAH”。然而,大多數蛋白質含有數百甚至數千個氨基酸,而不僅僅是三個!
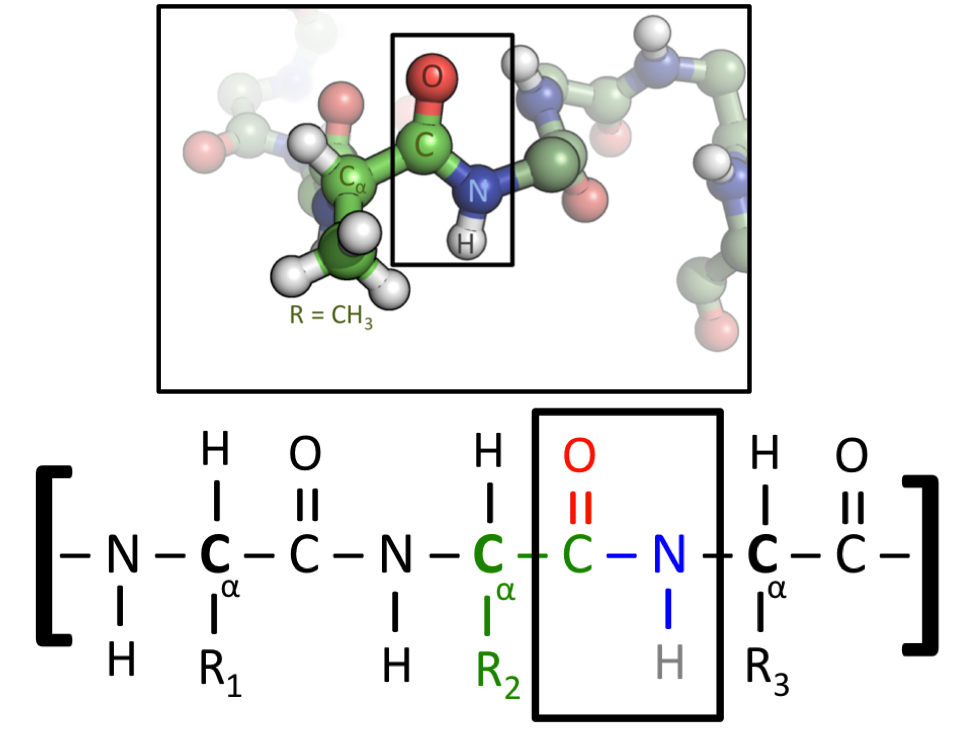
此圖顯示了蛋白質的兩種表示方式。所有氨基酸都包含碳-碳-氮序列。當氨基酸融合形成蛋白質時,這種重複模式將貫穿其整個長度,並被稱為蛋白質的“骨架”。然而,氨基酸在它們的“側鏈”上有所不同,側鏈是附著在碳-碳-氮骨架上的原子的名稱。下圖使用標記為R1、R2和R3的通用側鏈,它們可以是任何氨基酸。在上圖中,中心氨基酸有一個CH3側鏈——這表明它是氨基酸丙氨酸,它由字母A表示。(圖片來源)
儘管我們可以將它們寫成文字字串,但蛋白質實際上並不是一種“語言”,至少不是喬姆斯基會認可的任何一種語言。但是它們確實具有一些類似語言的特徵,從機器學習的角度來看,這使得它們與文字領域非常相似:蛋白質是由固定的小字母表中的長字串組成,理論上任何字串都是可能的,但在實踐中,只有非常小的一部分字串才真正“有意義”。隨機文字是垃圾,隨機蛋白質只是一個無定形的團塊。
此外,如果僅孤立地考慮蛋白質的某些部分,資訊就會丟失,就像僅閱讀從較長文字中提取的單個句子時資訊會丟失一樣。蛋白質的一個區域只有在存在其他能穩定和校正其形狀的蛋白質部分時,才能呈現其自然形狀!這意味著長程相互作用,即可以透過全域性自注意力很好地捕獲的相互作用,對於正確建模蛋白質非常重要。
至此,希望您對蛋白質是什麼以及生物學家為何如此關心它們有了一個模糊的認識——儘管它們的氨基酸“字母表”很小,但它們具有巨大的結構和功能多樣性,能夠僅僅透過檢視原始氨基酸“字串”來理解和預測這些結構和功能,將是一個極有價值的研究工具。
整合:蛋白質機器學習
現在我們已經瞭解了語言模型的遷移學習如何工作,以及蛋白質是什麼。有了這些背景知識,下一步就不難了——我們可以將相同的遷移學習思想應用於蛋白質!我們不再在涉及英語文字的任務上預訓練模型,而是在輸入是蛋白質但訓練資料充足的任務上訓練它。一旦完成,我們的模型有望學習到大量關於蛋白質結構的資訊,就像語言模型學習到大量關於語言結構的資訊一樣。這使得預訓練的蛋白質模型成為任何其他基於蛋白質的任務的理想選擇!
生物學家對訓練蛋白質模型感興趣的機器學習任務有哪些?最著名的蛋白質建模任務是蛋白質摺疊。這裡的任務是,給定氨基酸鏈,例如“MLKNV…”,預測該蛋白質最終會摺疊成的形狀。這是一項極其重要的任務,因為準確預測蛋白質的形狀和結構可以為蛋白質的功能及其運作方式提供大量見解。
早在現代機器學習出現之前,人們就開始研究這個問題——Folding@Home 等最早的大規模分散式計算專案,利用原子級別的模擬,以令人難以置信的空間和時間解析度來模擬蛋白質摺疊,並且有一個完整的蛋白質晶體學領域,利用 X 射線衍射觀察從活細胞中分離出來的蛋白質結構。
然而,與許多其他領域一樣,深度學習的到來改變了一切。AlphaFold,尤其是AlphaFold2,利用Transformer深度學習模型,並添加了許多蛋白質特有的功能,在僅憑原始氨基酸序列預測新型蛋白質結構方面取得了卓越的成果。如果您對蛋白質摺疊感興趣,我們強烈建議您檢視我們的ESMFold筆記本——ESMFold是一個類似於AlphaFold2的新模型,但它更像是一個“純粹”的深度學習模型,執行不需要任何外部資料庫或搜尋步驟。因此,設定過程要簡單得多,模型執行速度也快得多,同時仍保持出色的準確性。
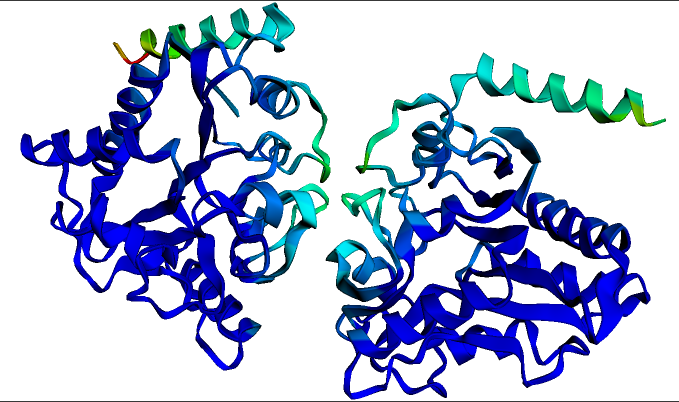
預測的同型二聚體多殺巴氏桿菌蛋白質**葡萄糖胺-6-磷酸脫氨酶**的結構。此結構和視覺化是使用上面連結的ESMFold筆記本在幾秒鐘內生成的。深藍色表示結構置信度最高的區域。
然而,蛋白質摺疊並非唯一令人感興趣的任務!生物學家可能希望透過蛋白質完成各種分類任務——也許他們想預測該蛋白質將在細胞的哪個部分發揮作用,或者蛋白質生成後,蛋白質中的哪些氨基酸會接受特定的修飾。用機器學習的語言來說,當您想要對整個蛋白質進行分類(例如,預測其亞細胞定位)時,此類任務稱為序列分類;當您想要對每個氨基酸進行分類(例如,預測哪些單個氨基酸會接受翻譯後修飾)時,則稱為標記分類。
然而,關鍵的啟示是,儘管蛋白質與語言截然不同,但它們幾乎可以透過完全相同的機器學習方法來處理——在大型蛋白質序列資料庫上進行大規模預訓練,然後將**遷移學習**應用於各種感興趣的任務,而這些任務的訓練資料可能稀疏得多。事實上,在某些方面它甚至比BERT等大型語言模型更簡單,因為不需要複雜的詞語分割和解析——蛋白質沒有“詞”的劃分,因此最簡單的方法是直接將每個氨基酸轉換為一個單獨的輸入令牌。
聽起來很酷,但我不知道從何開始!
如果您已經熟悉深度學習,那麼您會發現微調蛋白質模型的程式碼與微調語言模型的程式碼非常相似。如果您好奇,我們提供了PyTorch和TensorFlow的示例筆記本,您可以從像UniProt這樣的開放訪問蛋白質資料庫中獲取大量帶註釋的資料,該資料庫具有REST API和良好的網頁介面。您主要的困難將是找到有趣的探索性研究方向,這超出了本文件的範圍——但我相信有許多生物學家會樂於與您合作!
另一方面,如果您是生物學家,您可能已經有了一些想要嘗試的想法,但可能對深入研究機器學習程式碼有點望而卻步。別擔心!我們設計了示例筆記本(PyTorch,TensorFlow),以便資料載入部分與其他部分完全獨立。這意味著,如果您有一個**序列分類**或**標記分類**任務,您所需要做的就是構建一個蛋白質序列列表和相應的標籤列表,然後用您載入或生成這些列表的程式碼替換我們的資料載入程式碼即可。
雖然連結的具體示例使用ESM-2作為基礎預訓練模型,因為它代表了當前最先進的水平,但該領域的人們可能也熟悉Rost實驗室的模型,例如ProtBERT(論文連結),這些模型是同類模型中最早的一些,並引起了生物資訊學界的極大興趣。連結示例中的大部分程式碼可以透過簡單地將檢查點路徑從facebook/esm2...更改為Rostlab/prot_bert等內容,從而切換到使用ProtBERT等基礎模型。
總結
深度學習與生物學的交叉領域在未來幾年將是一個極其活躍且富有成果的領域。然而,深度學習之所以如此快速發展,其中一個原因在於人們能夠快速復現結果併為己所用。本著這種精神,如果您訓練了一個您認為對社群有用的模型,請務必分享!上面連結的筆記本包含將模型上傳到Hub的程式碼,其他研究人員可以在Hub上免費訪問和利用這些模型——除了對該領域的益處之外,這還是提高您的相關論文知名度和引用量的絕佳方式。您甚至可以使用Spaces製作一個即時網路演示,以便其他研究人員可以輸入蛋白質序列並免費獲取結果,而無需編寫任何程式碼。祝您好運,願審稿人2對您仁慈!







{kind=link}